Access the SynBioCAD Portal at Galaxy | SynBioCAD. Create your account, log in, and get started.
TOOLS: Important Tools can be quickly accessed using the Tools Panel located on the left side. You can search for your Favourite or any other tools using the search box;
- Get Data: This tool may be used to upload any file either from your computer or from a URL to your Galaxy workspace. This will add the file to the History section whose path can be defined in the desired workflows further.
-------------------------------------------------------------------------------------------
- SynBioCAD Utilities: Understanding the Utilities of the platform allows you to perform different tasks that enable you to create input files for the metabolic pathway generator tool called RetroPath2.0.
Source refers to the Target compound desired to be synthesized. This Galaxy tool uses the Source Compound Name and InChI string to construct a CSV file that can be used as the Source input file for RetroPath2.0.

Figure 1: Make source node output.
Sink refers to the metabolites of the chassis strain in which the Source needs to be produced. This tool uses an SBML file of the desired chassis organism’s dataset, parses all the species within it and uses the MIRIAM (1) annotations of the chemical species within a given compartment of the chassis organism (e.g. cytoplasm, Golgi apparatus, nucleus, etc) to find their InChI structures. You can use ‘Remove dead-end metabolites using FVA evaluation’ to conduct Flux variability analysis for removing metabolites that lack the requisite reactions (either metabolic or transport) that would account for their production or consumption within the metabolic network.
In the advanced options, one can specify the compartment from which the tool will extract the chemical species. The default is MNXC3, the MetaNetX code for the cytoplasm. If the user wishes to upload an SBML file from another source, then this value must be changed.
The results are written to a RetroPath2.0 friendly CSV file format that can be used as Sink input.
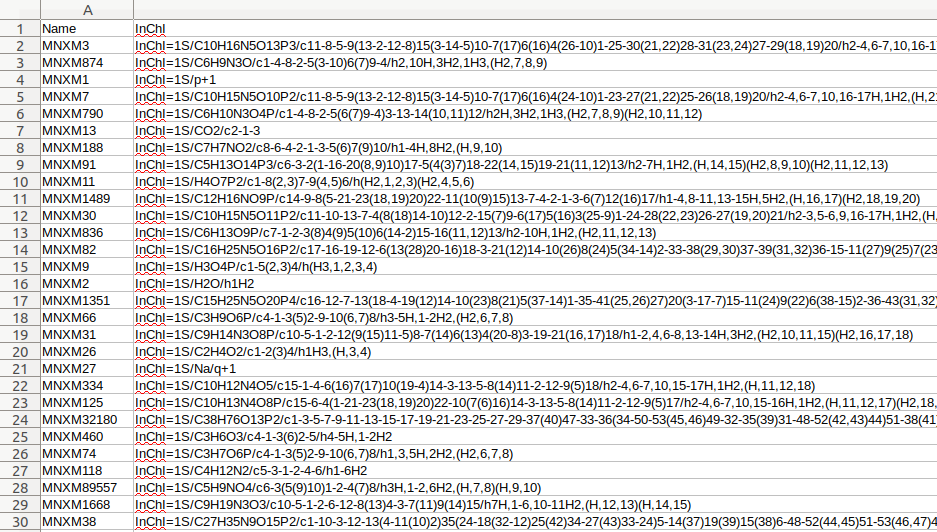
Figure 2: Example of output of the Sink from SBML.
References:
- Le Novère, Nicolas, et al. "Minimum information requested in the annotation of biochemical models (MIRIAM)." Nature Biotechnology 23.12 (2005): 1509-1515.
This tool uses the MIRIAM cross-references of the chemical species to identify similar species and merges only those that cannot be identified. The tool also uses species with in-house annotations that contain chemical structure descriptions (InChI, InChIkey, and SMILES) to compare species. Thereafter, it compares the reactants and products of the reactions and ignores the reactions that have the same reactants and products.
Extract Taxonomy: Extracts the taxonomy ID from an SBML
Extracts the taxonomic ID of the organism described in a given SBML (1).
References:
- Federhen, Scott. "The NCBI taxonomy database." Nucleic acids research 40.D1 (2012): D136-D143.
Given a collection or a single SBML file, this tool generates a CSV file output with all pathway information from the analysis tools (thermodynamics, FBA, etc). You can also specify the heterologous reactions and chemical species by providing the name of the heterologous pathway or SBML file ID.
It provides a graphical output of the heterologous pathway and the calculated pathway characteristics.
The CSV file generated contains a list of links to the DNA registry SynBioHub for the desired vector backbones, resistance cassette, and promoters.

Figure 3: An example of the OptDoE Genetic Parts Reference file generated.
-------------------------------------------------------------------------------------------
- SynBioCAD RetroSynthesis
- RetroRules: Generates reaction rules to use as input for RetroPath2.0
RetroRules: This tool retrieves and generates a list of reaction rules from the RetroRules database, which is in a RetroPath2.0 friendly format.
References:
Tool: https://retrorules.org/
RetroPath2.0 Docker: This tool performs retrosynthesis search for possible metabolic routes between a source molecule and a collection of sink molecules. It takes as input, files from RetroRules, source and sink compounds (can be generated as explained in SynBioCAD Utilities), and the maximal pathway length (i.e; the number of steps). Only a single source molecule is processed at this time.
Using Advanced Options, you can define the maximum number of compounds to keep for a next iteration (TopX), the minimal (dmin) and maximal (dmax) diameter for the reaction rules, and the maximum molecular weight of the source.
The Server URL is the IP address of the tools. By default, a local address is mentioned but external servers may be specified here too. Lastly, the Timeout parameter can be used to increase the maximum execution time of the tool. This limit is set for the server version of the tool. Another internal limit is set for the RAM usage upon deployment. This is designed to limit the potential combinatorial explosion that may occur with RetroPath2.0 and enable the tool to be deployed on a server for numerous users to access it simultaneously.
The output of the tool is a CSV file that describes the connection between the source and sink compounds through their different metabolic routes.
References:
RP2paths: RP2paths extracts the set of pathways that lies in a metabolic space file output by the RetroPath2.0 workflow. The advanced options include a “time out” parameter in ‘seconds’ to limit the execution time of the tool. Similar to the RetroPath2.0 node, an internal limit is set on RAM usage for this server version of the tool. The server URL is the IP address of the REST service tool, here localhost on port number 5008.
References:
Tool: https://github.com/brsynth/rp2paths
- Pathways to SBML: From the output of RP2Paths and RetroPath2.0, generates SBML pathways with mono-component reactions
This tool takes for input the outputs of both RetroPath2.0 and RP2paths and generates a series of SBML files with mono-component reactions.
Since more than one reaction rule may be associated with a single reaction; while converting to SBML, the best scoring reaction steps are considered before combining them to individual pathways. The number of combinations is controlled by the “out_paths max number of substeps per step” parameter in the Advanced Options. The default value is 2 but may be restricted to a minimum of 1 with no maximum. A higher value may be included to grow the potential size of the different pathways and is indicated in the filename as the last digit (ex: rp_1_1.sbml and rp_1_2.sbml indicates that these two models have the same reaction rules but not the same reactions associated with each). Users can use this parameter as per their suitability, but a combinatorial explosion may occur if a very high value is specified, meaning many rules are associated with many steps in a pathway.
The SBML compartment ID determines the name of the compartment in which the pathway will be added to, with a default of “MNXC3” that corresponds to the cytoplasm for MetaNetX models. The “rpSBML pathway ID” parameter is the ID of the groups (1) containing the heterologous reactions (default: rp_pathway), and the ID of the group containing the chemical species (default: central_species). Similarly, due to the creation of new reactions, the flux bounds need to be specified (default: irreversible reaction with a lower bound of 0 and an upper bound of 99999). Lastly, for those species that do not have cross-references to other databases, the tool provides the user an ability to query the PubChem REST web service using the InChIKey to retrieve more information.
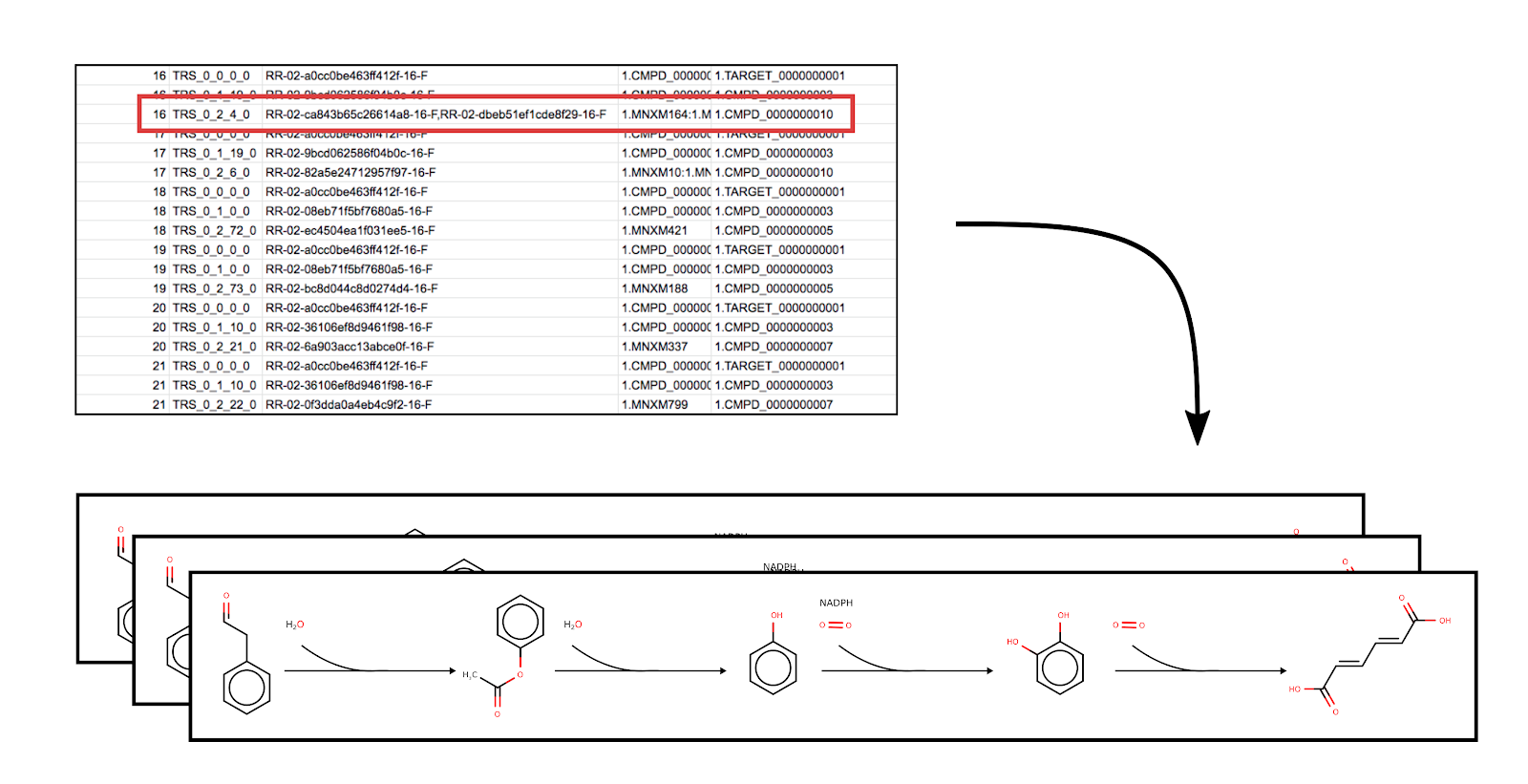
Figure 4: Illustration of Pathways to SBML converting the output of RP2paths to a collection of SBML files with the heterologous pathways stored within. Highlighted in red is a case where multiple rules are associated with a single reaction. Under such a condition, the tool calculates all the possible sub-paths of the pathway.
References:
- Hucka, Michael, and Lucian P. Smith. "SBML Level 3 package: Groups, Version 1 Release 1." Journal of integrative bioinformatics 13.3 (2016): 8-29.
Given the mono-component reactions generated by the tool “Pathways to SBML”, this node adds the cofactors associated with each mono-component reaction generated by RetroPath2.0. The advanced parameters include the name of the heterologous pathway defined by ‘Pathways to SBML’ node, the compartment ID of the SBML file (default is MNXC3, the MetaNetX ID for the cytoplasm). The tool also updates the reaction rule to include the SMILES cofactors to the reaction rule.
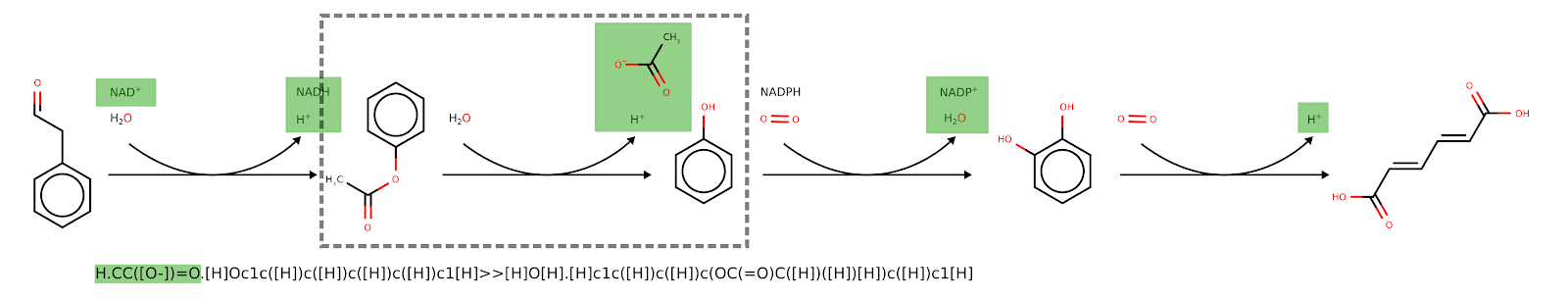
Figure 4: Example of cofactors (green) being added to a pathway calculated by RetroPath2.0. In the dashed box is an example of a reaction with its reaction rule with the added cofactors (green) added to the rule itself when these are added to the SBML file.
-------------------------------------------------------------------------------------------
- SynBioCAD Pathway Analysis
- Thermodynamics: Calculates the thermodynamic feasibility; i.e., the formation energy of chemical species, and the Gibbs free energy of their reactions and pathways in an SBML
This tool uses the component contribution (1) method to calculate the formation energy of chemical species, using InChI structures as well as MetaNetX and KEGG IDs. This method integrates multiple sources of information into a consistent framework that obeys the laws of thermodynamics and provides a significant improvement in accuracy compared to previous genome-wide estimations of standard Gibbs energies. Thereafter, the Gibbs free energy of reactions, and thus of the heterologous pathway is calculated. The tool requires the ID of the heterologous pathway to be specified.
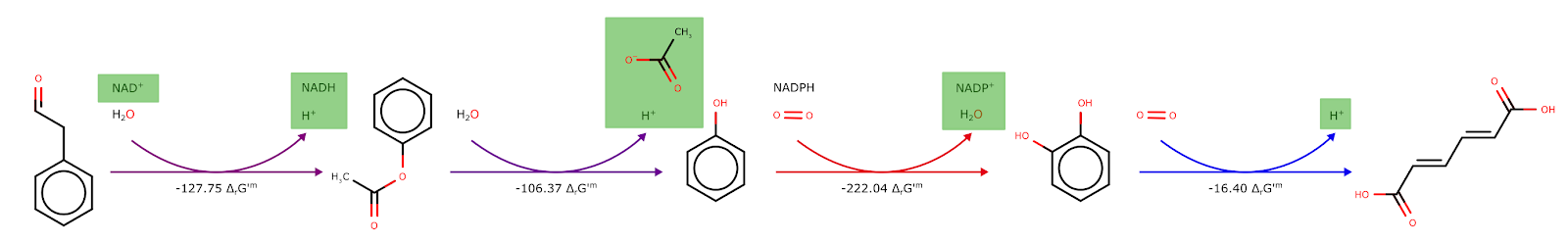
Figure 6: Illustration of the Gibbs free energy calculation using the component contribution package. The color-coded arrows from red (best) to blue (worst) show the best and worst-performing reactions.
References:
- Noor, Elad, et al. "Consistent estimation of Gibbs energy using component contributions." PLoS Comput Biol 9.7 (2013): e1003098.
Publication: eQuilibrator—the biochemical thermodynamics calculator | Nucleic Acids Research
Tool: eQuilibrator: The Biochemical Thermodynamics Calculator
Flux balance analysis is a mathematical approach for analyzing the flow of metabolites through a metabolic network. It is performed for heterologous pathways generated by RetroPath2.0. The tool performs the following steps:
- It merges a user-defined GEM SBML model with each given heterologous pathway individually.
- It performs FBA using the cobrapy (1) package. Three different analysis methods are proposed; two of which are native CobraPy methods - standard FBA and Parsimonious FBA, the other one proposed is an in-house analysis method named ‘Fraction of Reaction’.
For the simulation type ‘Fraction of Reaction’, this involves performing FBA using the ‘Source Reaction’ as the Objective function (by default the ‘biomass’ reaction is specified, which refers to the rate at which all of the biomass precursors are made in the correct proportions). Then the flux of that reaction has its upper and lower bounds set to the same value, determined as a ‘Fraction of the source reaction’ (default is 75% of its optimum). Thereafter, the objective is set to the target reaction (default setting is RP1_sink) followed by performing FBA once again. The tool uses the FBC package to manage the objective and flux bounds.
For the first two, the user must specify the name(s) of reaction(s) that the model will optimize to, while for the latter the user must provide the target reaction but also another source reaction that will be restricted.
Using the Advanced Options, the user can specify the name of the heterologous pathway as created by ‘Pathways to SBML’ and the compartment ID where the heterologous pathway exists in. The user may obtain a merged version of the resulting model, or the heterologous pathway only using the “Don’t output the merged model” boolean parameter. Using the ‘Maximize the Objective?’, you can choose if you’d like to maximize or minimize the objective (biomass production in this case) in the model.
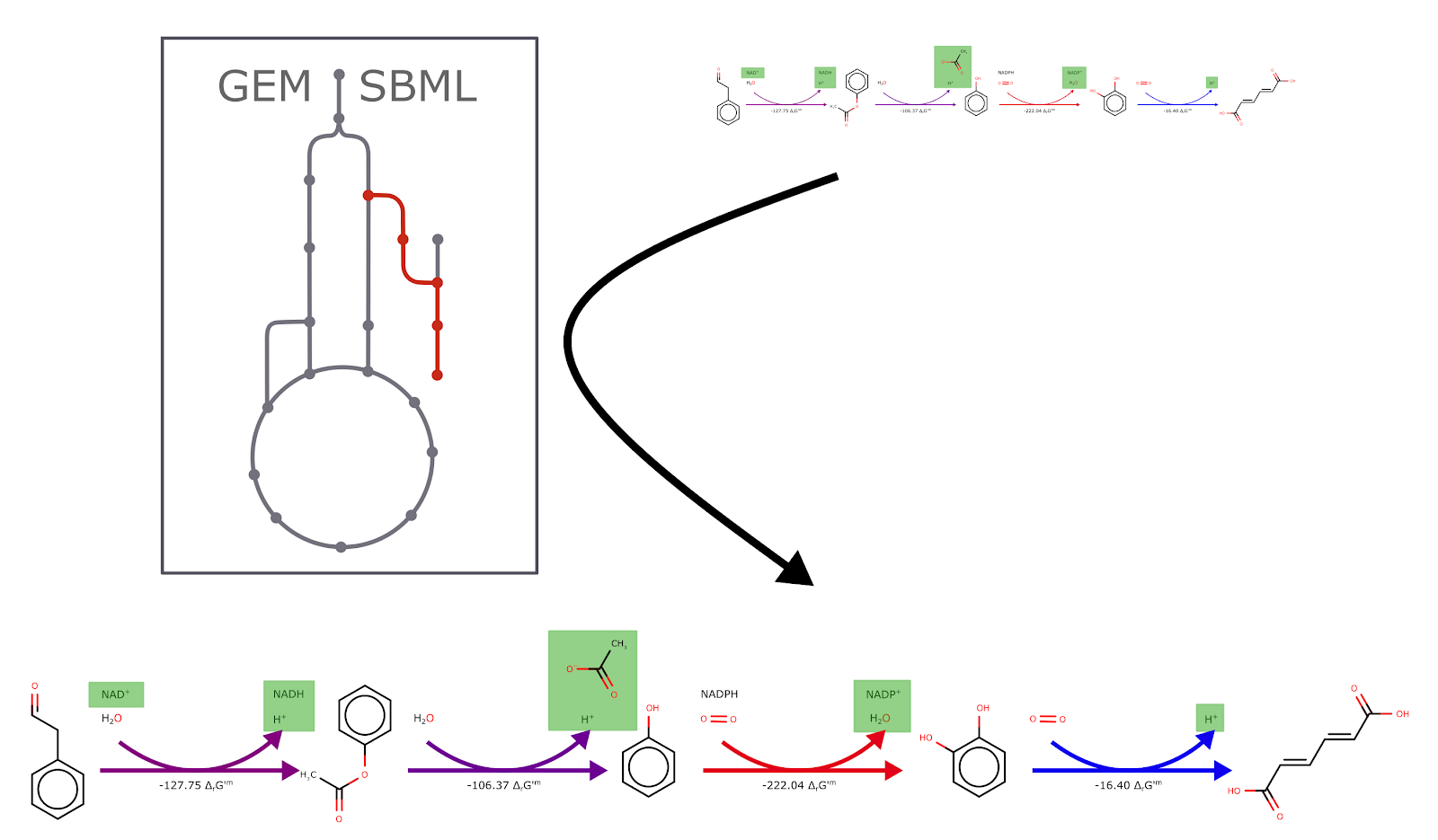
Figure 7: Illustration of the steps in the tool's calculation of FBA. The pathway is merged with a GEM SBML model and using the FBC package and CobraPy FBA is performed and the fluxes saved to the SBML file. The arrow size shows the individual step fluxes.
References:
- Noor, Elad, et al. "Consistent estimation of Gibbs energy using component contributions." PLoS Comput Biol 9.7 (2013): e1003098.
Paper: https://doi.org/10.1186/1752-0509-7-74
Tool: Documentation for COBRApy — cobra 0.13.3 documentation
- Rank Pathways: Calculates the global score based on thermodynamics, FBA, length, and reaction rule scores of a heterologous pathway
After performing all the different types of analysis as previously described, this tool normalizes the different values from each heterologous pathway, and performs a weighted sum function using the following attributes:
- Maximal number of steps in the pathway (as mentioned in RetroPath2.0 node, smaller pathways are better)
- Number of best scoring pathways/ models to return (Top Ranking Pathways)
As we do not know what is the importance of one result over another, each of these characteristics along with FBA and Thermodynamics has a weight associated with it that must be defined by the user between 0.0 and 1.0 and combined using a weighted sum function.
----------------------------------------------------------------------------------------------------
5. SynBioCAD Genetic Design
Using this tool, you can also define the characteristics of the Ribosome Binding Site (RBS) at this stage, the corresponding sequence of which will be added later using the PastGenie node.
References:
the Synthetic Biology Open Language (SBOL) | a language for genetic design
- Selenzyme: Reads the reaction rule SMILES from the collection of rpSBML files and queries Selenzyme for the associated enzymes and their similarity scores.
In each predicted Pathway, for each reaction, the tool takes for input a tar.xz with a collection of rpSBML files or a single rpSBML file (containing a reaction rule SMILES for each heterologous reaction), scans for the reaction rules, makes a REST request to Selenzyme, and finds corresponding enzymes (through Uniprot ID's) catalyzing all the reactions in heterologous pathways of rpSBML files.
Using the Advanced Options, you can assess similarities between enzymes by specifying the Chemical fingerprint as ‘RDK’, ‘Pattern’, or ‘Morgan’(1).
References:
Tool: http://selenzyme.synbiochem.co.uk
This tool takes as input a TAR collection of rpSBML files, that contain for each heterologous reaction a ranked list of UniProt identifiers for the sequences, and the list of links to the DNA registry SynBioHub for the desired vector backbones, resistance cassette, and promoters (generated using the tool ‘OptDoE Parts Reference Generator’). An optimal design of experiments is performed by using OptBioDes based on logistic regression analysis with an assumed linear model for the response. The design is evaluated by its D-efficiency, defined as:

Where n is the number of experimental runs or library size, p is the number of independent variables, and X is the model matrix, i.e., a row for each experimental run and a column for each term in the model. The library size n can be selected and should be above a minimal threshold depending on the number of combinatorial complexity of the library.
The experimental design can be evaluated through the provided diagnostics for D-efficiency, power analysis, and relative prediction variance, allowing the optimal selection of library size.
The resulting design is provided as an SBOL collection containing the definition of each DNA component and the combinatorial library of constructs.
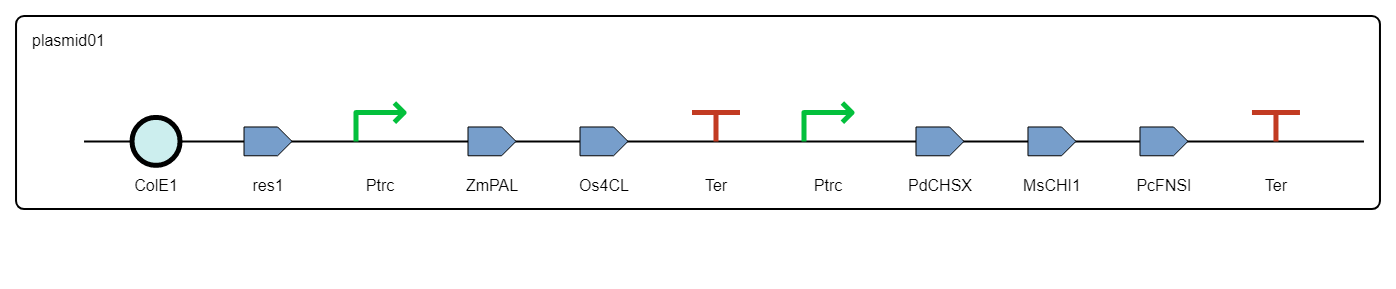
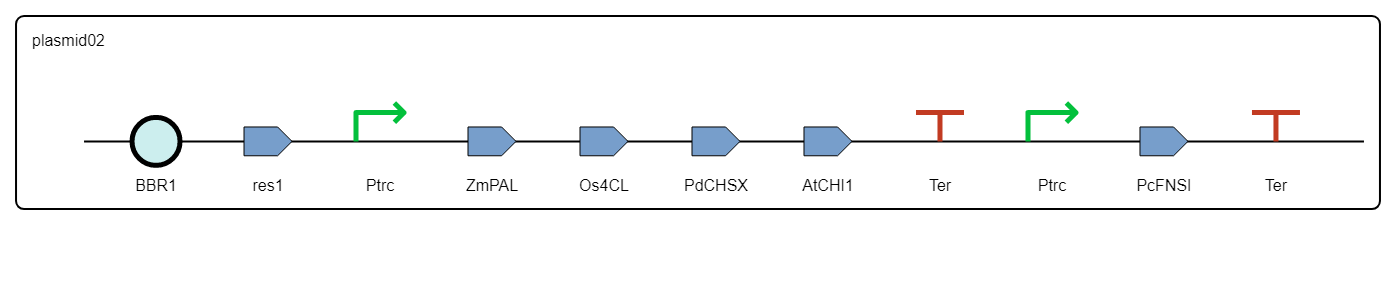
Figure 8. Example of two constructs generated by rpOptBioDes through optimal combinatorial design and represented using SBOL Visual in SynBioHub.
PartsGenie: PartsGenie allows for the design and optimization of novel, reusable synthetic biology parts.
For each enzyme predicted, it recovers the DNA sequence from the UNIPROT ID, runs a codon optimization protocol, and adds various strength calculated RBS (ribosome binding site) to it.
Reference:
Tool: PartsGenie
- DNA Weaver: Given an SBOL input, it calculates assembly parts for Gibson or Golden Gate
This tool uses a multi-objective algorithm to predict the best synthesis and assembling strategy for the previously designed plasmid using either Golden gate assembly, or Gibson assembly, or a mix of both. Given a set of designs (one design is a construct name and list of its parts), it finds a valid and efficient assembly plan to build all the designs. The designs and sequences of parts are provided as an SBOL file (see test_input.xml for an example)
Method:
- We assume that the different standard parts are available or will be ordered, with the exact sequence provided in the input file (in the future it would be easy to automatically break long parts into smaller fragments).
- The desired construct sequence for a genetic part design is simply the concatenation of that design's part sequences in the right order (no assembly overhang is included).
- Buy primers with overhangs to extend the parts fragments via PCR and create homologies between them so they can be assembled together.
- Assemble each construct in a single step with Golden Gate assembly if possible (that is if at least one site out of BsaI, BbsI, and BsmBI is totally absent from the construct sequence), else with Gibson assembly. It can also be only one of the two methods if the option Gibson or golden_gate is selected instead of any_method.
- Start with the first design, and for each subsequence design assembly plan reuse the primers ordered and fragments PCRed in previous designs, if relevant.
Here is a schema of the supply network used:
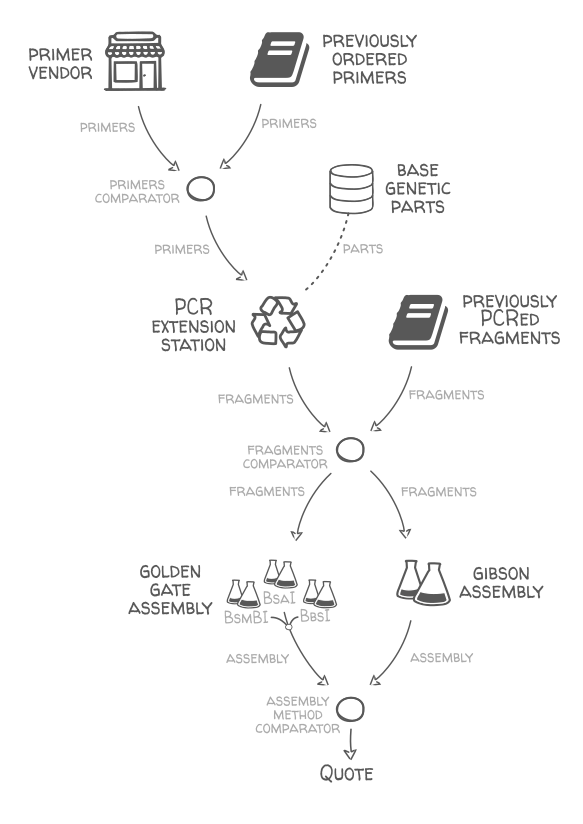
Output:
See example_output.xlsx for an example. The output is an Excel spreadsheet with the following sub-sheets:
- construct_parts: the ID and list of part names (in the right order) for each design.
- construct_sequences: the final sequence of the constructs to build.
- part_sequences: the list of each standard part and its sequence (same information as in the input SBOL file).
- fragment_extensions: for each PCR fragment, the standard part and the primers to use
- assembly_plan: for each design, the list of PCR fragments to use.
- errors: list of errors to help troubleshooting assemblies for which no valid assembly plan was found.
Description of the example/testing sample:
The example input SBOL is from an example file provided by @pablocarb, with a random sequence used for the terminator (Ter) part. The example has 48 designs that are well represented. Some have type-2s sites preventing the golden gate, some use the same part more than once, making it a challenging scenario for scarless Golden Gate assembly.
The output example_output.xlsx shows the plan generated to build all the designs. The plan has:
- 19 different base parts (provided by the SBOL input)
- 125 primers to be ordered to extend the parts in various ways (that's less than 3 primers per design to build, thanks to primer reuse)
- 116 fragments to be PCRed (less than 3 PCRs per design to build, thanks to fragment reuse).
- 15 designs seem to need Gibson assembly as they contain BsaI, BsmBI, and BbsI sites, the rest use Golden Gate assembly.
Limitations
For constructs with repeated parts and other homologies (such as, in the example, the designs with several "Ter" in a row, ), Gibson assembly (and probably LCR assembly too) may create mis-annealed constructs and more clones will need to be picked. This is not taken into account by the script at the moment. This could be fixed by buying custom fragments from a commercial vendor for extreme cases (i.e. by amending the current implementation to forbid Gibson cuts in regions with homologies elsewhere and add a DNA vendor in the supply network).
Written by Zulko at the Edinburgh Genome Foundry.
Reference:
Tool: https://edinburgh-genome-foundry.github.io/DnaWeaver
- LCR Genie: Given an SBOL, calculate assembly parts for ligase cycling reaction.
The ligase chain reaction (LCR) is a method of DNA amplification that differs from PCR in that it involves a thermostable ligase to join two probes or other molecules together which can then be amplified by standard PCR cycling.