Name: Block:
PCR Amplification of DNA[1]
The polymerase chain reaction (PCR) is a DNA amplification technique that has revolutionized almost all aspects of biological research. The PCR technique was invented in 1984 by Dr. Kary Mullis while at Cetus Corporation. Mullis was awarded a Nobel Prize for his work in 1994. PCR allows for the amplification of a small quantity of DNA over one million-fold. The enormous utility of PCR is based on its procedural simplicity and specificity. Since the first application of PCR to diagnose sickle cell anemia, a large number of procedures have been developed. PCR has made amplification of DNA an alternate approach to cloning experiments. PCR is also used extensively in forensics, paternity/kinship testing, and the identification of human remains.
PCR amplification requires the use of a thermostable DNA polymerase. The most commonly used of these is Taq DNA polymerase, purified from a bacterium known as Thermus Aquaticus that inhabits hot springs. This enzyme remains stable at near-boiling temperatures. Also included in the PCR reaction are the four deoxynucleotides (dATP, dCTP, dGTP, and dTTP) and two synthetic oligonucleotides, typically 15-30 base pairs in length, known as "primers". These components, together with the DNA to be amplified, are incubated in an appropriate buffer that contains Mg2+. The primers are designed and synthesized to correspond to the start and end of the DNA to be amplified, known as the "template" or "target". If the template DNA is prepared from biological tissue, freshly isolated DNA will give the best amplification results. DNA extracted from older specimens may be degraded and therefore less suitable for amplification.
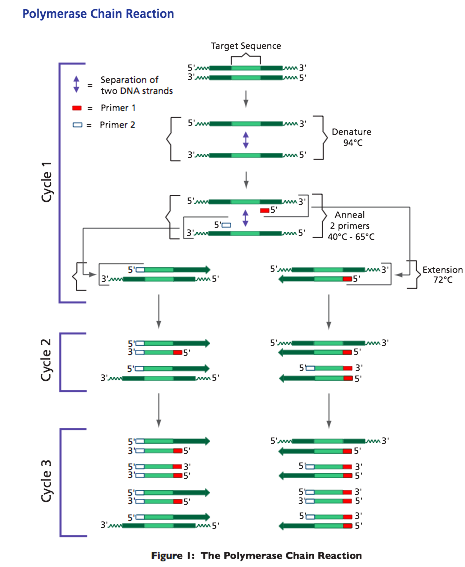
The PCR reaction mixture (which contains the Taq DNA polymerase, buffer, deoxynucleotides, primers, and template) is subjected to sequential heating/cooling cycles at three different temperatures. The three temperatures are the basis of the PCR process (Figure 1). In the first step, the template is heated to near boiling (92° - 96°C.) to denature or "melt" the DNA. This step, known as "denaturation" disrupts the hydrogen bonds between the two complementary DNA strands and causes their complete separation. In the second PCR step, the mixture is cooled to a temperature that is typically in the range of 45° - 65°. In this step, known as "annealing", the primers, present in great excess to the template, bind to the separated DNA strands. In the third PCR step, known as "extension", the temperature is raised to an intermediate value, usually 72°C. At this temperature the Taq DNA polymerase is maximally active and adds nucleotides to the primers to complete the synthesis of the new complementary strands.
The exact temperature and incubation time required for each step depends on several factors, including the length of the target DNA and GC content of the primer/template. In some cases, the annealing and extension steps may be combined resulting in a two step per PCR cycle.
The three PCR steps of denaturation, annealing, and extension constitute one "cycle" and result in a doubling of the template copies in the mix- ture. The process is typically repeated for 20-40 cycles. Theoretically, if the reaction is allowed to be repeated for (n) cycles, the number of copies of template DNA will be 2n following completion, as shown at the bottom of Figure 1. For example, one would anticipate one million-fold amplification after 20 cycles. In theory, this process could continue indefinitely. In practice, however, the amount of product reaches a maximum after about 40 cycles, due to the depletion of reaction components and loss of DNA polymerase activity.
One common problem that occurs during PCR is unwanted amplification products. These are due to contamination of the sample or nonspecific annealing of the primers. To reduce contamination, autoclaved tubes, pipet tips, and sterile water should be used. Gloves should always be worn when performing PCR.
To minimize unwanted PCR products due to nonspecific primer annealing, the primer concentration should be minimized, if possible. Another common technique is "hot start", in which the components of the PCR reaction are fully mixed only after the DNA is fully denatured above 94°C.
Following PCR, the amplified product is processed, depending on the objective of the experiment. In most cases, the DNA is subjected to either aga- rose or polyacrylamide gel electrophoresis. In DNA fingerprinting, used in criminal forensics, PCR-amplified DNA from the crime scene is compared to amplified DNA from suspects. In cloning experiments, the amplified DNA is typically further purified and ligated into the desired vector. In DNA sequencing experiments, the amplified DNA (usually radioactively labeled) is run on a thin polyacrylamide gel sequencing gel. The gel is then placed on a piece of film, which is activated by the radioactive molecules and developed to create an image of the DNA fragments in the gel. Alternatively, one may avoid the use of radioactivity by subjecting the sequencing gel to silver staining, in which silver ions bind directly to the DNA fragments.
EXPERIMENT OBJECTIVE:
The objective of this experiment is for students to gain hands-on experience of the principles and practice of Polymerase Chain Reaction (PCR).
This experiment has three modules:
- PCR Reaction
- Separation of PCR Reactions by Electrophoresis
- Size Determination of PCR Amplified DNA Fragment
Module 1: PCR Reaction Using a Thermal Cycler
- Label three 0.5 ml tubes: "0" (Control), "15", and "30" and with your initials or group designation.
Control Reaction:
- To tube “0” (control), add the following:
- 2 μl DNA Template for Amplification
- 2 μl Primer Mix
- 5 μl 10x gel loading solution
- 10 μl Enzyme Grade Ultrapure Water
PCR Reaction
- Transfer the PCR Reaction Pellet to a 0.2 ml tube.
- Label the tube containing the PCR Reaction Pellet as "PCR" and with your initials or group number.
- Tap the PCR reaction tube to assure that the PCR reaction pellet is at the bottom of the tube. Add the following to the PCR tube:
- 10 μl Primer Mix (two primers)
- 7 μl Enzyme Grade Ultrapure Water
- 10 μl DNA Template for Amplification
- Gently mix the reaction tube and shake to ensure all material is at the bottom of the tube.
Polymerase Chain Reaction Cycling
- Program the thermal cycler for a total of 30 cycles. Each cycle will be:
- 94°C for 45 seconds
- 45°C for 45 seconds
- 72°C for 45 seconds
On the final cycle, the 72°C incubation can be extended to 5 minutes.
- After the 15th cycle, remove 7 μl of the PCR mixture and transfer the sample to the tube you earlier labeled "15".
- Add 5 μl of 10x Gel Loading solution and 8 μl of Enzyme Grade Water to the 7 μl sample in the tube labeled "15". Store on ice until ready for electrophoresis.
- Return the “PCR” tube to the thermal cycler and continue the PCR reaction to the 30th cycle.
- After the 30th cycle, remove 7 μl of the PCR mixture and transfer the sample to the tube you earlier labeled "30".
- Add 5 μl of 10x Gel Loading solution and 8 μl of Enzyme Grade Water to the 7 μl sample in the tube labeled "30". Store on ice until ready for electrophoresis the following class.
Module II: Separation of PCR Reactions by Agarose Gel Electrophoresis
Preparing the Agarose Gel
- Close off the open ends of a 7x7cm clean and dry gel bed (casting tray) by using rubber dams or tape.
- Place a well-former template (comb) in the first set of notches at the end of the bed. Make sure the comb sits firmly and evenly across the bed.
- To a 250 ml erlenmeyer flask , add 0.2 g of agarose powder, 0.5 ml of 50x buffer and 24.5 ml distilled water. Swirl the mixture to disperse clumps of agarose powder.
- With a marking pen, indicate the level of the solution volume on the outside of the flask.
- Heat the mixture using a microwave oven to dissolve the agarose powder; microwave for batches of 45 seconds each until liquid is completely translucent.
- Cool the agarose solution to 60°C with careful swirling to promote even dissipation of heat. If detectable evaporation has occurred, add distilled water to bring the solution up to the original volume marked in step 4.
After the gel is cooled to 60 C
- Place the bed on a level surface and pour the cooled agarose solution into the bed.
- Allow the gel to completely solidify. It will become firm and cool to the touch after approximately 15 minutes.
- After the gel is solidified, be careful not to damage or tear the wells while removing the rubber dams or tape and comb(s) from the gel bed.
- Place the gel (on its bed) into the electrophoresis chamber, properly oriented, centered and level on the platform.
- Fill the electrophoresis apparatus chamber with the appropriate amount of diluted (1x) electrophoresis buffer to completely cover/submerge the gel.
Loading DNA Samples
- Heat the DNA Standard marker and PCR samples for two minutes at 50°C. Allow the samples to cool for a few minutes.
- Make sure the gel is completely submerged under buffer before loading the samples. Load 20 μl each of the samples in the following sequence:
Lane | Tube | |
1 | Marker | Standard DNA Fragments |
2 | 0 | Control reaction, 0 cycle |
3 | 15 | Reaction sample, 15 cycles |
4 | 30 | Reaction sample, 30 cycles |
- Record the position of your sample in the gel for easy identification after staining.
Running the Gel
- After the DNA samples are loaded, properly orient the cover and care- fully snap it onto the electrode terminals
- Insert the plugs of the black and red leads into the corresponding inputs of the power source.
- Set the power source to approximately 125 volts and conduct electrophoresis for 25 minutes.
- Check to see that current is flowing properly, you should see bubbles forming on the two platinum electrodes.
- After the electrophoresis is completed, disconnect the power and remove the gel from the bed for staining.
Staining and Visualization of DNA
- Remove the 7 x 7 cm agarose gel from its bed and completely submerge the gel in a small, clean tray containing 75 ml of distilled or deionized water, or used electrophoresis buffer. The agarose gel should be completely covered with liquid.
- Gently float a 7 x 7 cm card of InstaStain® Blue with the stain side (blue) facing the liquid.
- Let the gel soak undisturbed in the liquid for approximately 3 hours. The gel can be left in the liquid overnight (cover with plastic wrap to prevent evaporation).
- After staining and destaining, the gel is ready for visualization and photography.
Module III: Size Determination of PCR Amplified DNA Fragment
The size of the PCR amplified DNA fragment can be extrapolated by its migration distance relative to the Standard DNA Fragments, for which the size of each fragment is known.
- Measure and record the distance traveled in the agarose gel by each Standard DNA fragment (except the largest 23,130 bp fragment, which will not fit in a straight line in step 4).
In each case, measure from the lower edge of the sample well to the lower end of each band. Record the distance traveled in centimeters (to the nearest millimeter).
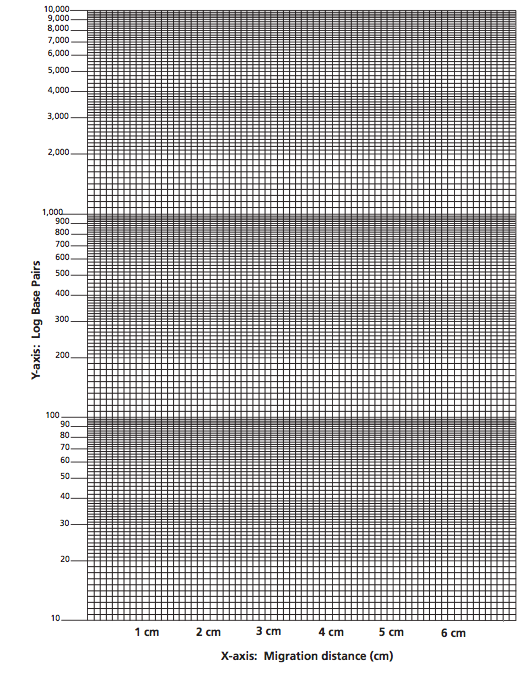
- Label the semi-log graph paper:
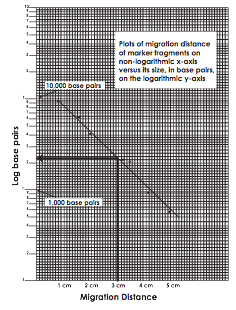
- Label the non-logarithmic horizontal x- axis "Migration Distance" in centimeters at equal intervals.
- Label the logarithmic vertical y-axis "Log base pairs". Choose your scales so that the data points are well spread out. Assume the first cycle on the y-axis represents 100-1,000 base pairs and the second cycle represents 1,000-10,000 base pairs.
- For measured migration distance on the x-axis versus its size in base pairs, on the y-axis.
- Draw the best average straight line through all the points. The line should have approximately equal numbers of points scattered on each side of the line. Some points may be right on the line (see Example figure at left).
- Using the graph of the Standard DNA fragments, determine the sizes in base pairs of the main PCR amplified DNA from this curve.
- Find the migration distance of the PCR amplified DNA on the x-axis. Draw a vertical line from that point until the standard graph line is intersected.
- From the point of intersection, draw a second line horizontally to the y-axis and determine the approximate size of the PCR amplified DNA in base pairs (refer to Figure example).
[1] Lab adapted from Edvotek PCR Amplification of DNA Lab #330